MAXRD SYMBOL
ImportCrystalData
ImportCrystalData[cif]
extracts information from a cif file and stores the data in $CrystalData under the cif file name.
ImportCrystalData[cif,label]
extracts information from a cif file and stores the data in $CrystalData under label.
ImportCrystalData[{label,formula,Z,sg,λ},lattice,atomData,notes]
stores the data in $CrystalData under label.
ImportCrystalData[]
opens a dialogue for entering the data.
Details and Options
- If data is added manually, notes may be omitted. It can be either zero or any number of strings. Each element entry should should be an Association with the following keys: "Element", "FractionalCoordinates", "DisplacementParameters", (ADPs) "Type", and notes if wanted.
- ImportCrystalData will overwrite the data file containing the crystal data. The default file is seen from the default setting of "DataFile".
- Options include:
-
"DataFile" FileNameJoin[{$MaXrdPath,"UserData","CrystalData.m"}] path to data file "ExtractSubdata" 1 part to extract in cif files containing multiple structures "Notes" <> notes or comments to the data "OverwriteWarning" True whether to prompt user with warning dialogue "RoundAnglesThreshold" 0.001 angles within this threshold will be rounded to integers "Units" True whether to store numbers as a Quantity - If the function fails to find any space group information, a message appears prompting the user to enter that information manually. If the user cancels (or enters an empty string), the first space group (
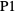 ) will be used.
) will be used. - If the .cif file does not contain information concerning the atomic displacement parameter type (_atom_site_adp_type in coreCIF dictionary), displacement parameters will be set to zero and the type to "Uiso". The following table gives and overview of the type (left column) and the content of the "DisplacementParameters" entry (right column):
-
Uani 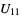 ,
, 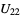 ,
, 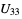 ,
, 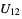 ,
, 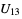 and
and 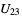
Uiso 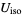 (or
(or 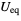 )
)Bani 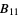 ,
, 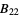 ,
, 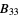 ,
, 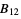 ,
, 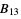 and
and 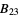
Biso 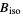 (or
(or 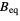 )
) - Space groups will be stored as formatted symbols unless it corresponds to an alternative setting, in which case the full Hermann–Mauguin symbol will be used (see ToStandardSetting).
- Certain non-crucial data elements will not be stored. For example, if all occupation factors are one or all displacement parameters are zero, those keys will be deleted from the "AtomData" “section”.
- The following data items are searched for in the .cif files:
-
lattice parameters: cell lengths _cell_length_a
_cell_length_b
_cell_length_clattice parameters: angles _cell_angle_alpha
_cell_angle_beta
_cell_angle_gammaatom data: fractional coordinates _atom_site_fract_x
_atom_site_fract_y
_atom_site_fract_zatom data: occupation factor _atom_site_occupancy atom data: site symmetry multiplicity/order _atom_site_site_symmetry_multiplicity
_atom_site_site_symmetry_orderatom data: ADPs (anisotropic) _atom_site_aniso_U_11
_atom_site_aniso_U_22
_atom_site_aniso_U_33
_atom_site_aniso_U_12
_atom_site_aniso_U_13
_atom_site_aniso_U_23atom data: ADPs (isotropic) _atom_site_U_iso_or_equiv atom data: ADP type _atom_site_adp_type radiation wavelength _diffrn_radiation_wavelength formula units ( 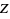 )
)_cell_formula_units_Z chemical formula _chemical_formula_iupac
_chemical_formula_structural
_chemical_formula_sumspace group _space_group_name_Hall
_space_group_name_H-M_alt
_space_group_IT_number
_symmetry_space_group_name_Hall
_symmetry_space_group_name_H-M
_symmetry_Int_Tables_number - The function will extract the space group from the following CIF labels (in prioritised order):
_space_group_name_Hall
_space_group_name_H-M_alt
_space_group_IT_number
_symmetry_space_group_name_Hall
_symmetry_space_group_name_H-M
_symmetry_Int_Tables_number - If data is found on one of the labels, it will be run through InputCheck with the "InterpretSpaceGroup" label.
- The function will extract the chemical formula from the following CIF labels (in prioritised order):
_chemical_formula_iupac
_chemical_formula_structural
_chemical_formula_sum
If none of them are found, the user is prompted with a window to enter the formula manually. Also, formulas containing commas or question marks will be ignored. - To remove entries from $CrystalData, one can use KeyDropFrom[$CrystalData,crystal].

